

Химийн урвалын явцад молекулууд нь тухайн урвал явагдах зайлшгүй нөхцөл болох шилжилтийн төлөвт хүрэх хүртэл харилцан үйлчлэлээр энерги хуримтлуулдаг. Энэ төлөв нь богино хугацаанд явагддаг тул туршилтаар ажиглахад хүндрэлтэй юм. Эдгээр шилжилтийн төлөвийг квант химийн аргуудаар тооцоолж болох ч тооцоолол нь маш их цаг хугацаа шаарддаг. MIT -ийн судалгааны баг шилжилтийн төлөвийг богино хугацаанд тооцоолж чадах тооцооллын аргыг машин сургалтын арга дээр суурилан хөгжүүлсэн байна. Энэхүү шинэ аргыг түлш эсвэл эмийн молекул гэх мэт ашигтай бүтээгдэхүүн гарган авах, шинэ урвал, катализатор зохион бүтээх, байгальд тохиолддог химийн урвалыг загварчлахад ашиглаж болно. “Шилжилтийн төлөв дэх молекулын бүтцийг мэдэх нь катализаторыг загварчлахад эсвэл байгаль дээр төлөвийн шилжилт хэрхэн явагддаг болохыг ойлгоход чухал юм.” гэж тус судалгааны удирдагч тайлбарлажээ.
Аливаа химийн урвал явагдахын тулд шилжилтийн төлөвийг дамжих ёстой ба энэ нь урвал явагдахад шаардлагатай энергийн босгод хүрэх үед тохиолддог байна. Химийн урвал явагдах магадлал нь шилжилтийн төлөв үүсэх магадлалаар тодорхойлогдоно. “Шилжилтийн төлөв нь химийн хувирал тохиолдох магадлалыг тодорхойлоход тусалдаг. Хэрэв бидэнд нүүрстөрөгчийн давхар исэл гэх мэт зүйлс байгаад түүнийг метанол гэх мэт ашигтай түлш болгох хувилгахыг хүсэж байгаа бол шилжилтийн төлөв хэр өндөр магадлалтай байх нь бидний урвалжаас бүтээгдэхүүн хэр хэмжээгээр гарган авах магадлалыг тодорхойлдог.” гэж өгүүллийн зохиогчдын нэг тайлбарлажээ.
Химичид нягтын функцийн онол гэж нэрлэдэг квант химийн аргыг ашиглан төлөвийн шилжилтийг тооцоолж байна. Гэхдээ энэ арга нь их хэмжээний тооцооллын хүчид чадал шаарддаг бөгөөд нэг шилжилтийн төлөвийг тооцоолохын тулд олон цаг зарцуулдаг байна. Сүүлийн үед зарим судлаачид шилжилтийн төлөвийн бүтцийг илрүүлэхийн тулд машин сургалтыг аргыг ашиглахыг оролдож байна. Гэхдээ одоог хүртэл хөгжүүлсэн аргууд нь хоёр урвалжийг нэгнээсээ хамааралтай орон зайн байрлалтай нэг объект гэж үздэг байна. Нэгнээсээ хамааралгүй боломжит байрлалуудыг тусдаа урвал гэж үздэг бөгөөд энэ нь тооцооллын хугацааг нэмэгдүүлдэг. “Хэрэв урвалд орж буй молекулууд хөдөлгөөнтэй бол зарчмын хувьд эргэлдэх үедээ ижил химийн урвалд олж болно. Гэхдээ машин сургалтын арга дахь сургалтын загвар нь эдгээрийг хоёр өөр урвал гэж үзэх болно. Энэ нь машин сургалтын сургалтыг хүндрүүлж, нарийвчлалыг бууруулдаг” гэж зохиогч тайлбарлажээ. Тус судалгааны баг уусмалын диффузын загвар гэж нэрлэдэг загварыг ашиглан хоёр урвалж нэгнээсээ хамааран чөлөөтэй байрлах боломж олгодог тооцооллын шинэ аргыг хөгжүүлсэн бөгөөд энэ арга нь урвалын процессын аль төрөл нь урвалын бүтээгдэхүүн үүсэх магадлалыг өндөр болохыг тогтоох болно. Эдгээр загварыг сургах сургалтын өгөгдөлдөө квант тооцооллын аргыг ашиглан гарган авсан 9000 ялгаатай химийн урвалын урвалжийн төлөв, бүтээгдэхүүний төлөв болон урвалжуудаас бүтээгдэхүүн үүсэхэд дамжсан шилжилтийн төлөвийн бүтцийг ашигласан байна. “Сургалтын загвар эдгээр гурван төлөв хэрхэн зэрэгцэн оршдогийг мэдсэний дараа бид тус сургалтын загварт шинэ урвалжууд болон бүтээгдэхүүнийг өгч болох бөгөөд загвар нь эдгээр шинэ урвалж болон түүний бүтээгдэхүүнтэй холбогдох шилжилтийн төлөвийн бүтцийг таамаглахыг оролдох болно.” гэж өгүүллийн холбоо барих зохиогч тайлбарлажээ. Тус судлаачид өөрсдийн сургалтын загвараа өмнө нь тооцоолж байгаагүй 1000 урвал дээр туршсан ба шилжилтийн төлөв бүрийн хувьд 40 боломжит бүтэц гарган авсан байна. Дараа нь тэд аль төлөв тохиолдох магадлал хамгийн өндөр болохыг таамаглахын тулд үнэлгээний “итгэлцлийн загвар” ашигласан байна. Тус сургалтаар гарган авсан бүтцүүд нь квант химийн тооцооллоос гарсан шилжилтийн төлөвийн бүтэцтэй харьцуулахад 0.08 Å нарийвчлалтай байсан байна. Урвал бүрийн хувьд сургалтын тооцоолол нь хэдхэн секунд хугацааг зарцуулсан байна. Хэрэв мянган шилжилтийн төлөвийг квант тооцооллын аргаар тооцоолно гэвэл хэр их цаг хугацаа шаардахыг хэлэхэд хэцүү юм. Хэдийгээр тус судлаачид загвараа ихэвчлэн цөөн атом агуулсан нэгдлүүдтэй холбоотой урвал дээр сургасан ч том молекулууд оролцох урвалын хувьд өндөр нарийвчлалтай таамаглаж болохыг тогтоосон байна.
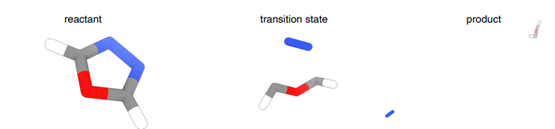
Зураг: Энгийн урвал
Одоо тус судлаачид катализатор гэх мэт бусад нэгдлийг оруулж өгөхийн тулд загвараа өргөжүүлэхээр ажиллаж байгаа бөгөөд энэ нь катализатор урвалыг хэрхэн хурдасгадаг болохыг судлах, ойлгох, тайлбарлахад тусална гэж найдаж байгаа юм. Энэ нь эмийн бүтээгдэхүүн, түлшний бүтээгдэхүүн, болон бусад ашигтай нэгдлүүдийг гарган авах шинэ процессыг боловсруулахад ашигтай байж болох юм. Энэ төрлийн загварын өөр нэг боломжит хэрэглээ нь бусад гаригаас олдсон хий хоорондын тохиолдож болох харилцан үйлчлэлийг судлах эсвэл дэлхий дээрх амьдралын хувьслын эхэн үед тохиолдсон байж болох энгийн химийн урвалыг загварчлахад ашиглаж болох юм гэж тус судлаачид хэлжээ.
Илүү дэлгэрэнгүй мэдээллийг Chenru Duan, Yuanqi Du, Haojun Jia, Heather J. Kulik. Accurate transition state generation with an object-aware equivariant elementary reaction diffusion model. Nature Computational Science, 2023; DOI: 10.1038/s43588-023-00563-7, өгүүллээс уншина уу.
Эх сурвалж: Computational model captures the elusive transition states of chemical reactions | ScienceDaily
Мэдээ бэлтгэсэн: Симуляци, тооцооллын салбар – ЭША М.Чагдаржав