
Молекулын бүтцийг загварчлах, динамикийг тооцоолох нь ямар үр ашигтай вэ?
Өнөөдрийн бидний хэрэглэж буй эм, материал, гоо сайхны бүтээгдэхүүн, хүнсний нэмэлт гэх мэт олон зүйл нь молекулын түвшинд маш нарийн бүтэц, харилцан үйлчлэлтэй байдаг. Эдгээр бүтцийг шууд нүдээр харах боломжгүй тул судлаачид цөмийн соронзон резонанс, рентген дифракц гэх мэт аргуудыг ашиглан ажигладаг. Гэхдээ эдгээр аргаар ажигласан бүтэц нь зөвхөн нэг агшин дахь атомуудын байрлал буюу тухайн тогтсон төлөвийг илэрхийлдэг. Харин бодит байдалд молекулууд нь байнга хөдөлгөөнд оршиж, харилцан үйлчлэлд ордог тул хэд хэдэн төлөвт оршин байдаг. Иймээс төлөв бүрийн бүтцийг туршилтаар тодорхойлох нь ихээхэн цаг хугацаа, зардал шаардсан ажил юм. Үүнийг зохицуулах, молекулын бүтэц, хөдөлгөөнийг нарийн судлах, сонирхолтой шийдэл нь молекулын бүтцийг загварчлах, динамикийг тооцоолох юм.
Молекулын динамик симуляци гэж нэрлэдэг арга нь хугацаанаас хамааруулан молекул систем дэх бүх атомын хөдөлгөөнийг физикийн хуулиуд дээр тулгуурлан тооцоолдог. Өөрөөр хэлбэл, энэ арга нь Ньютоны хөдөлгөөний тэгшитгэлийг ашиглан молекул систем доторх атом бүрийн байрлал, хурд, энергийн өөрчлөлтийг нарийн тооцдог байна. Симуляци хийснээр бид молекулууд хэрхэн хөдөлж, хэлбэлзэж, харилцан үйлчилж, ямар бүтэц үүсгэж байгааг ажиглаж болно. Жишээ нь, энэ симуляцийн арга нь уураг хэрхэн эвхрэхийг,[1] пептид хэрхэн угсрагдаж нано бүтэц үүсгэдгийг, [2] эмийн молекул уурагтай хэрхэн харилцан үйлчилж байгааг [3] компьютерт суурилан тооцоолох боломжийг олгосон. Одоо уншигч та “ийм симуляци хийх нь ямар үр ашигтай байдаг юм бол?” гэж бодож байна уу?
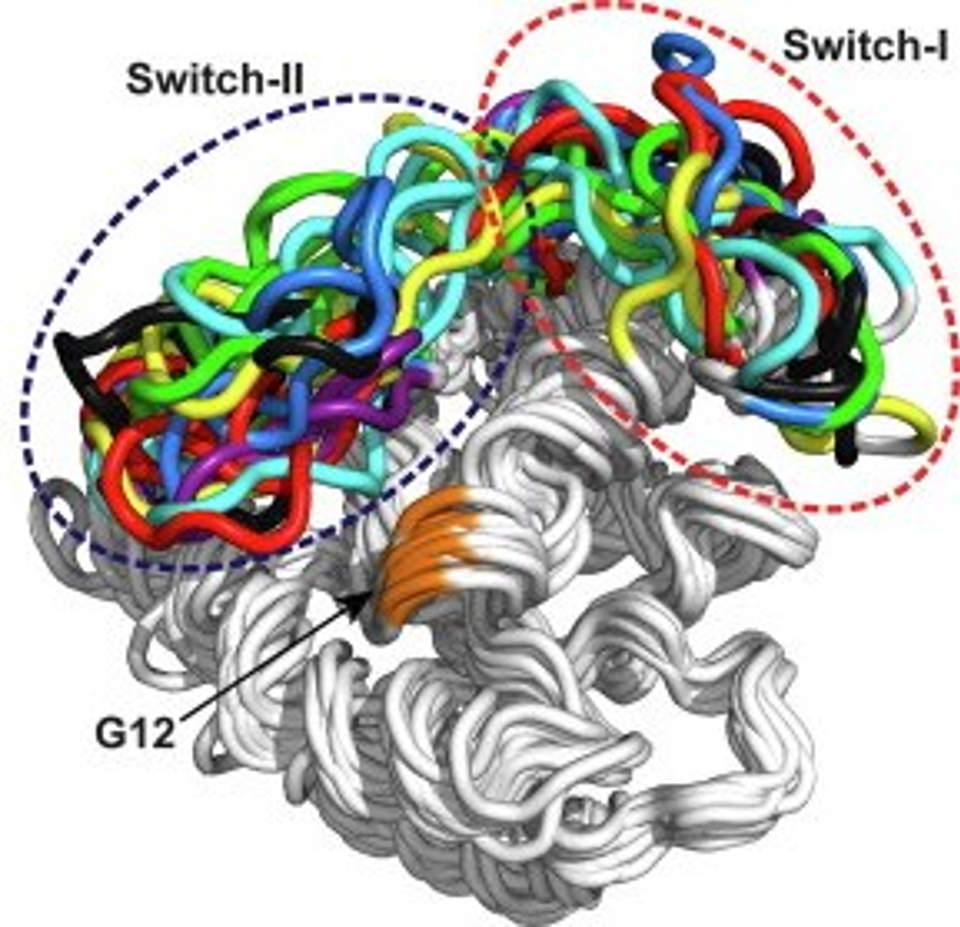
Молекулын загварчлал, динамик симуляцийн аргууд нь шинжлэх ухааны олон салбарт бодит туршилтыг орлох төдийгүй түүнийг гүнзгийрүүлэн ойлгох шинэ боломжийг нээж өгсөн байна. Жишээлбэл, эм зүйн судалгаанд молекулын түвшний тооцооллоор эмийн нэгдэл уургийн идэвхтэй хэсэгтэй хэрхэн холбогдож, ямар атом хоорондын харилцан үйлчлэл давамгайлж байгааг тодорхойлдог.[4] Ийм судалгаа нь туршилтаар олон жил шаардагдах процессыг богино хугацаанд, өндөр нарийвчлалтайгаар урьдчилан таамаглах боломж олгодог тул шинэ эмийн зохиомжийг хурдасгаж, зардлыг асар их хэмжээгээр бууруулдаг. Мөн уураг, пептидийн эвхрэх, өөрөө угсрагдах, нано бүтэц үүсэх үйл явц, уураг, уураг-РНХ эсвэл ДНХ, мембран болон бусад молекул хоорондын харилцан үйлчлэл зэрэг байгалийн нарийн механизмыг ажиглахад эдгээр аргыг өргөн ашигладаг. [5], [6], [7] Эдгээр судалгаагаар гарсан үр дүн нь өвчний шалтгааныг илрүүлэх,[8] эсийн мембраныг дуурайсан шинэ төрлийн биоматериал бүтээх, эсвэл эмийн хүргэлтийн ухаалаг систем боловсруулах,[9] зэрэг бодит хэрэглээнд хүрдэг.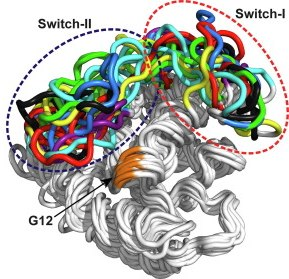
Зураг. KRAS уургийн бүтэц (хөдөлгөөн ихтэй хэсэг, G12 мутаци), T. Pantsar нар [8].
Материал судлалын салбарт молекулын түвшний симуляци нь бат бөх, хөнгөн, дулаан болон цахилгааныг сайн дамжуулах чадвартай шинэ материалын бүтцийг урьдчилан таамаглахад ашиглагддаг. Жишээлбэл, нүүрстөрөгчийн нано хоолой, графеныг ашиглан илүү уян хатан электрон төхөөрөмж, мэдрэгч, нанокомпозит материал боловсруулах боломжийг олгосон.[10] Мөн хүрээлэн буй орчны судалгаанд уусгагчийн динамик, бохирдуулагч бодисын молекулын түвшний тархалтыг тооцоолсноор байгаль орчны хор нөлөөг бууруулах арга замыг тодорхойлох боломжтой.
Молекулын динамик симуляци нь зөвхөн тоон өгөгдөл гаргах хэрэгсэл биш, харин байгалийн хууль, бодисын бүтэц, бодит үйл явцыг молекулын түвшинд “дүрслэн харуулах” дижитал лаборатори юм. Өнөөдөр дэлхийн тэргүүлэх их сургуулиуд, судалгааны хүрээлэнгүүд энэ аргыг ашиглан шинэ материал, ухаалаг эм, нанотехнологийн бүтээгдэхүүн, эрчим хүчний үр ашигтай шийдэл боловсруулахад өрсөлдөж байна.
Эх сурвалж:
[1] M. Karplus and G. A. Petsko, “Molecular dynamics simulations in biology,” Nature, vol. 347, no. 6294, pp. 631–639, Oct. 1990, doi: 10.1038/347631a0.
[2] B. Mijiddorj, H. Shirakata, T. Nakagawa, K. Ueda, Y. Yokoyama, and I. Kawamura, “Stereochemical Effects on the Self-Assembly of Pyrenylalanine-Phenylalanine Dipeptide,” Bulletin of the Chemical Society of Japan, vol. 93, no. 8, pp. 969–977, Aug. 2020, doi: 10.1246/bcsj.20190376.
[3] B. Bayarsaikhan, B. Z. Zsidó, R. Börzsei, and C. Hetényi, “Efficient Refinement of Complex Structures of Flexible Histone Peptides Using Post-Docking Molecular Dynamics Protocols,” IJMS, vol. 25, no. 11, p. 5945, May 2024, doi: 10.3390/ijms25115945.
[4] D. Al-Fahad, G. Ropón-Palacios, D. A. Omoboyowa, G. Singh, and R. B. Patil, “Virtual screening and molecular dynamics simulation of natural compounds as potential inhibitors of serine/threonine kinase 16 for anticancer drug discovery,” Mol Divers, vol. 29, no. 2, pp. 1525–1539, Apr. 2025, doi: 10.1007/s11030-024-10931-8.
[5] M. Honma and H. Suzuki, “Can molecular dynamics facilitate the design of protein–protein-interaction inhibitors?,” Nature Reviews Rheumatology, vol. 19, no. 1, pp. 8–9, Jan. 2023, doi: 10.1038/s41584-022-00877-2.
[6] Y. Zhang and M. Ding, “Probing nanopores: molecular dynamics insights into the mechanisms of DNA and protein translocation through solid-state and biological nanopores,” Soft Matter, vol. 21, no. 13, pp. 2385–2399, 2025, doi: 10.1039/D4SM01534G.
[7] D. Ham et al., “Conformational switch that induces GDP release from Gi,” Journal of Structural Biology, vol. 213, no. 1, p. 107694, Mar. 2021, doi: 10.1016/j.jsb.2020.107694.
[8] T. Pantsar, “The current understanding of KRAS protein structure and dynamics,” Computational and Structural Biotechnology Journal, vol. 18, pp. 189–198, 2020, doi: 10.1016/j.csbj.2019.12.004.
[9] M. Khalkhali, S. Mohammadinejad, F. Khoeini, and K. Rostamizadeh, “Vesicle-like structure of lipid-based nanoparticles as drug delivery system revealed by molecular dynamics simulations,” International Journal of Pharmaceutics, vol. 559, pp. 173–181, Mar. 2019, doi: 10.1016/j.ijpharm.2019.01.036.
[10] N. Chaudhary and M. K. Dikshit, “A state of art review on the graphene and carbon nanotube reinforced nanocomposites: A molecular dynamics approach,” Materials Today: Proceedings, vol. 47, pp. 3235–3241, Jan. 2021, doi: 10.1016/j.matpr.2021.06.440.
Мэдээ бэлтгэсэн: Симуляци, тооцооллын салбар ЭША- М.Чагдаржав.